
Molecular Dynamics
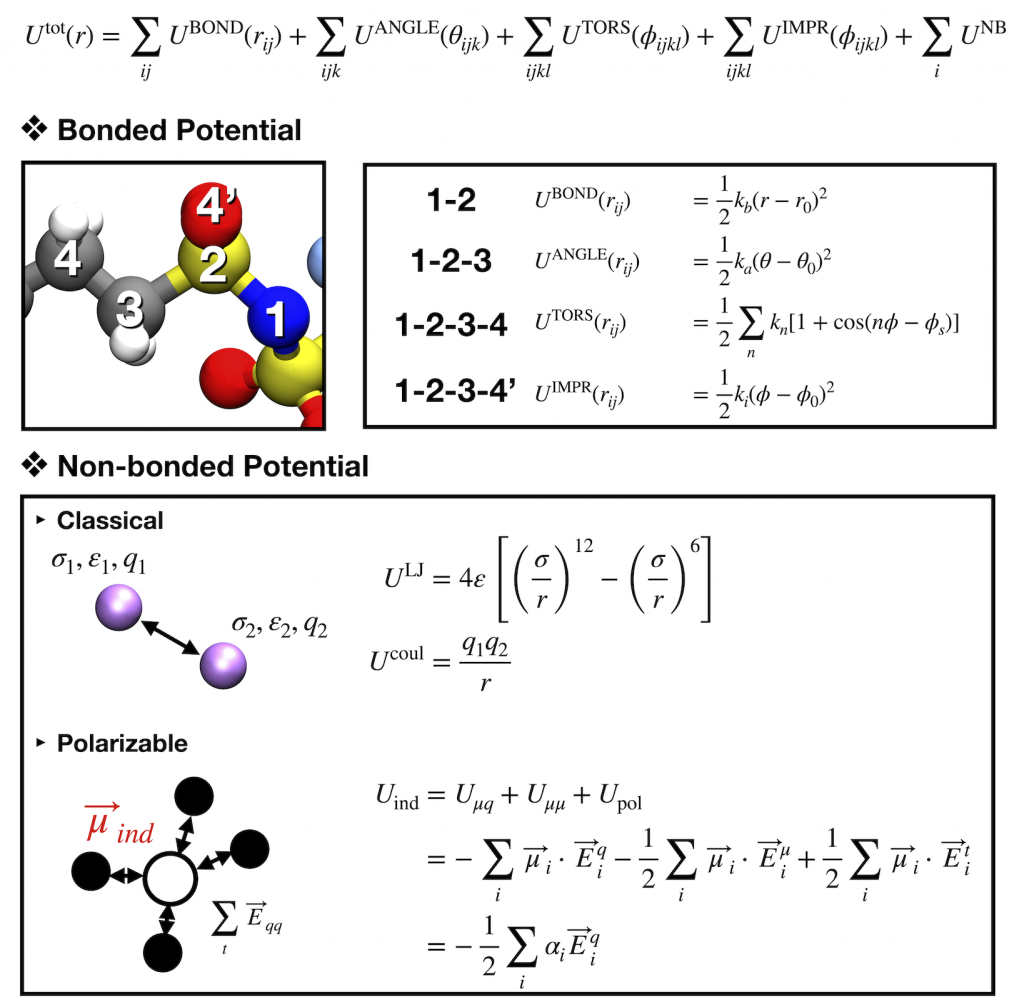
Molecular dynamics (MD) is a well-known tool to understand physical phenomena in condensed systems. Although the rapid development in computational resources allow the expansion in the research area of the first principle calculation such as DFT (density functional theory), MD simulation is still powerful to elucidate complex systems like bio-systems, polymers, and electrolytes for Li-ion batteries.
In contrast to other molecular simulation methods like Monte Carlo, the positions and velocities of each atom in the systems can be obtained along their trajectories based on the Newton dynamics. The positions and velocities are determined by the forces between particles or temperatures, and the set of the force parameters is called a force field [Left]. With proper force fields, structural and dynamic properties can be obtained for various systems. Here, the polarizable force field based on induced point dipole model was applied to investigate Li ion dynamics in polyelectrolytes or ionic liquids. With the induced dipoles calculated by local electric fields of each atom, more precise prediction for Li dynamics as well as interaction with other particles was possible than conventional MD simulations.
Li-ion Transport in Polymer Branched Nanopore with Ionic Liquids

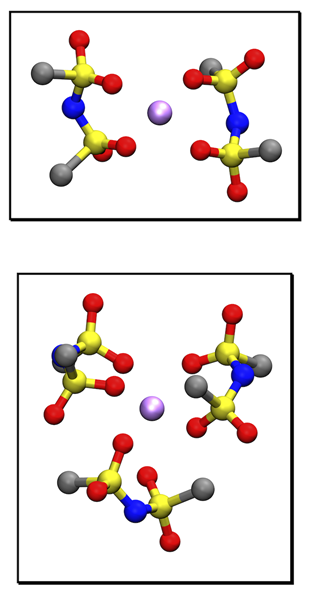
It is known that the dynamics of Li ions in ionic channels of the polyelectrolyte is enhanced by branched poly-(ethylene glycol) (PEO) chains [Anh, 2019]. In present study, thorough investigation in atomistic scale was conducted by polarizable molecular dynamics simulations. The analysis for local environment of Li ions and each ion cluster showed predominance of smaller cluster [LiTFSI2]- than [LiTFSI3]-2 which is more present in binary electrolytes without PEO polymers allowed Li ions to behave more structural diffusion in the electrolytes. In addition, the dynamics was more faster for the electrolytes with the higher salt concentration and the smaller diameter of channels.
Semi-IPN conductors with Several Additives
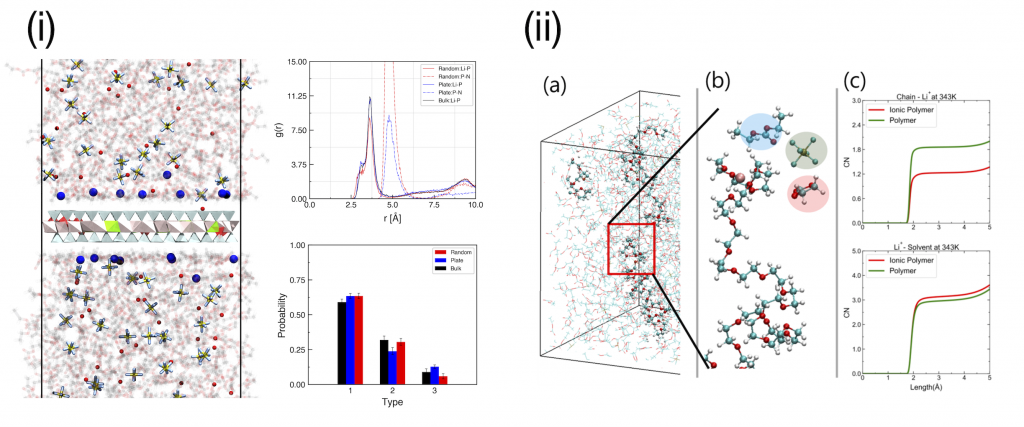
- Advanced Energy Materials 2020, 10, 47, 2003114
- Advanced Energy Materials 2021, 11, 44, 2102660
Several additives have been introduced to Li-ion batteries to improve not only electrical performance but also stability or safety. With inorganic clays (i) or polymer additives (ii), is was confirmed that enhanced performances of semi-IPN conductors by both experimental and computational results.
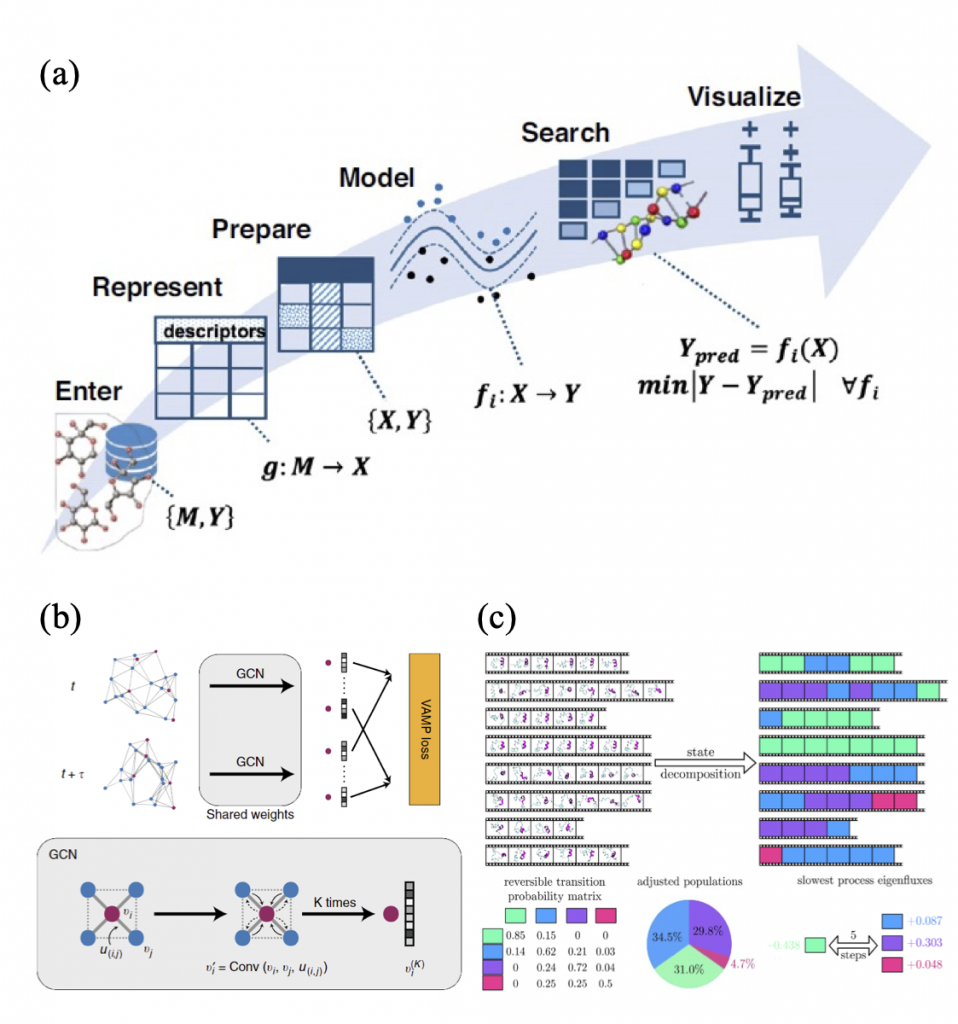
Machine Learning Approach in Li-ion Battery Systems
 - Current Opinion in Chemical Engineering 2019, 23:51–57
- Current Opinion in Chemical Engineering 2019, 23:51–57
- Nature Communications 2019, 10:2667
- Journal of the American Chemical Society, (2018), 2386-2396, 140(7)
Machine Learning (ML) is a data-derived prediction technique whose models are different from physics- based models. ML can extract complex and often hidden correlations (f) between features (X) and properties (Y) from given datasets, which are difficult to be obtained by conventional analysis methods. With these correlations, ML models can predict properties from newly given datasets. For chemistry and material science, the properties can be mechanical, structural, dynamical, etc.
If one analyzes MD simulation trajectories (time series data) and extract important correlations by ML methods, Graph Dynamical Networks (GDyNets) and Markov state models (MSMs) will be useful. The GDyNet is an unsupervised learning method for learning atomic scale dynamics from MD trajectories of arbitrary condensed phases. A GDyNet classifies local configurational states around a target atom using graph convolutional neural networks (GCN). To analyze local environment of Li ions in Li-ion battery electrolytes, one can assign Li ions as target atoms. Similarly, an MSM decomposes MD trajectories into a few states using clustering methods based on structural and kinetic similarities between molecular conformations.
Both ML methods classifies states where the Markovian assumption is satisfied, and then obtain a transition matrix whose components are transition probabilities between the states. From these states and transition probabilities, various structural and dynamic properties can be obtained: radial distribution functions, ionic conductivities, transition timescales, reactive fluxes, etc. From these properties, The Li- ion atomic dynamics and transport mechanism can be revealed.